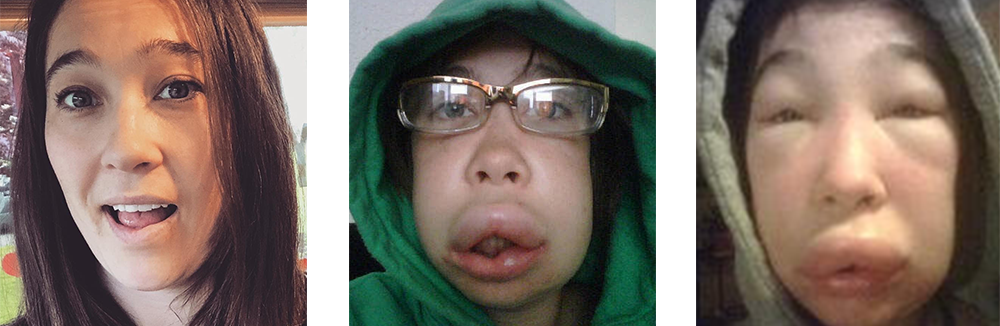
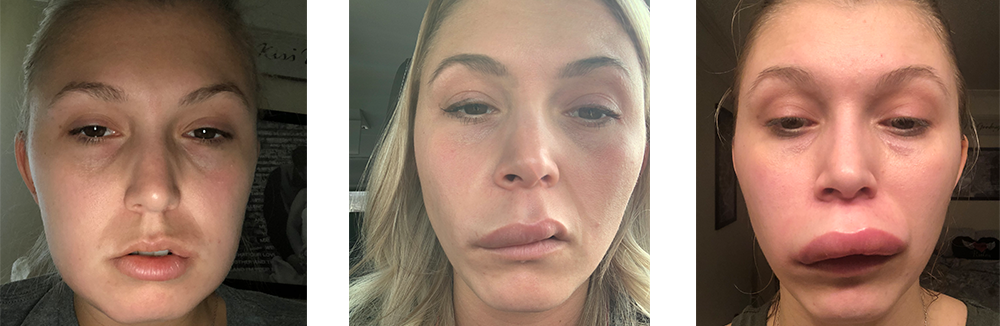
El Angioedema Hereditario (AEH), es una condición genética muy rara y potencialmente mortal que involucra ataques recurrentes de hinchazón severa (angioedema) en varias partes del cuerpo, incluidas las manos, los pies, los genitales, el estómago, la cara y / o la garganta. La hinchazón de las vías respiratorias puede restringir la respiración y ser fatal. Los episodios pueden desencadenarse por un trauma físico o estrés emocional; sin embargo, la hinchazón a menudo ocurre sin un desencadenante conocido.
Los síntomas del AEH suelen aparecer a temprana edad, con mayor frecuencia a los 13 años, y pueden aumentar en severidad después de la pubertad. Debido a que el AEH es tan poco común, puede llevar hasta una década en obtener un diagnóstico preciso después de que se experimentan los síntomas por primera vez.
Cuando no se trata, un ataque de AEH suele durar tres días, a veces incluso más, y muchas personas con AEH experimentan tres o más ataques de hinchazón por mes. La frecuencia y la gravedad de los ataques varían significativamente entre las personas, incluso entre los miembros afectados de la misma familia.
La gran mayoría de las personas con AEH tienen un defecto genético que causa una deficiencia en la proteína plasmática llamada Inhibidor-C1. El AEH también se observa en personas que tienen niveles normales de Inhibidor-C1, sin embargo, los defectos genéticos en otros genes causan su angioedema.
Las personas con AEH experimentan episodios recurrentes de hinchazón en las manos, los pies, los genitales, el estómago, la cara y / o la garganta que pueden durar de dos a cinco días. La frecuencia y la gravedad de los ataques pueden variar drásticamente en las personas afectadas por el AEH, incluso en las de la misma familia. Aproximadamente el 25% de las personas con AEH experimentan un sarpullido rojo plano y sin picazón que a menudo ocurre antes o durante un ataque de AEH.

La gran mayoría de las personas con AEH tienen un defecto en el gen que controla una proteína sanguínea llamada Inhibidor-C1. Este defecto provoca un desequilibrio bioquímico que produce hinchazón. AEH también se conoce como Deficiencia de Inhibidor-C1 Tipo I y Tipo II. Las personas diagnosticadas con AEH con Inhibidor-C1 Normal experimentan síntomas de hinchazón generalmente similares a los observados en los tipos I y II. La HAEA está financiando activamente investigaciones diseñadas para comprender mejor las causas genéticas y bioquímicas subyacentes y encontrar tratamientos para los miembros de nuestra comunidad que sufren de AEH con Inhibidor-C1 Normal.
El AEH es hereditario y los niños tienen un 50% de posibilidades de heredar el AEH si uno de los padres tiene la enfermedad. Sin embargo, la ausencia de antecedentes familiares no descarta el diagnóstico de AEH, ya que los informes científicos indican que hasta un 25% de los casos de AEH son el resultado de una mutación espontánea del gen de Inhibidor-C1 en el momento de la concepción. Los hijos de personas con AEH también pueden heredar la afección.
El AEH se considera una enfermedad muy rara y se presenta en aproximadamente 1 de cada 10,000 a 1 de cada 50,000 personas. El Tipo I es el más común y representa aproximadamente el 85 por ciento de los casos. El Tipo II ocurre en aproximadamente el 15 por ciento de los casos. No hay datos sobre la incidencia de AEH con Inhibidor-C1 Normal, sin embargo, los científicos creen que la población representa un pequeño subconjunto de personas que padecen AEH.

Si bien algunos ataques parecen ser espontáneos, algunas de las causas más comunes de los ataques de AEH son:
Las personas con AEH también han reportado hinchazón en las extremidades de la siguiente manera:

 HORMONAS
HORMONAS INHIBIDOR ACE
INHIBIDOR ACE PROCEDIMIENTOS DENTALES
PROCEDIMIENTOS DENTALES LA MAYORÍA de los casos de angioedema o hinchazón NO son deficiencia de AEH con Inhibidor-C1.
El AEH es muy poco común y la mayoría de las personas, incluidos los profesionales médicos, no están familiarizadas con la enfermedad hasta que se encuentran con alguien que la padece. Incluso, a medida que los síntomas van y vienen, es común que las personas con AEH permanezcan sin diagnosticar durante muchos años. El dolor abdominal frecuente y severo puede diagnosticarse erróneamente fácilmente, a veces incluso resultando en una cirugía exploratoria innecesaria.
El diagnóstico adecuado es fundamental para el tratamiento y el manejo exitoso del AEH. Se requieren análisis de laboratorio de muestras de sangre o muestras genéticas para establecer un diagnóstico de AEH. Hay tres análisis de sangre específicos que se utilizan para confirmar el Angioedema Hereditario Tipo I o II:
1. Inhibidor-C1 cuantitativo (antigénico)
2. Inhibidor-C1 Funcional
3. C4
Las pruebas genéticas para el AEH con Inhibidor-C1 Normal pueden determinar si hay un defecto en uno de los otros tres genes que se ha demostrado que también causan AEH.